
[서리풀 연구通] 국가의 책임을 묻다
연두 (시민건강연구소 회원)
지난 11월 26일 국제탐사보도언론인협회(ICJI)가 주도하여 36개국 59개 언론기관이 함께 글로벌 의료기기 산업의 문제점을 파헤친 결과가 3일에 걸쳐 <뉴스타파>에 보도 되었다. 의료기기의 허가 절차에 문제가 있다는 이야기가 가끔씩 언론에 보도된 적은 있지만, 이렇게 문제점들이 총체적으로, 적나라하게 드러난 것은 처음이었다. (☞관련 기사 : 연 450조 의료기기산업…결함 등으로 10년간 8만 명 사망).
고령의 대동막판막 협착증 환자가 수술 대신 받을 수 있는 고가의 시술에 사용되는 인공심장판막 제품에 안전성 문제가 있다는 사실, 의료기기에 문제가 발견되어 제조사가 이를 회수했음에도 한국 식품의약품안전처가 이 내용을 충분히 알리지 않았다는 사실, 의료기기를 사용하는 대형병원 의사들이 업체로부터 이런 저런 형태의 관리를 받고 있다는 사실에 이르기까지, 이번 보도가 담고 있는 내용은 놀라운 것이었다.
의료기기를 둘러싼 이런 문제들은 환자의 건강, 그것도 죽음 또는 그에 못지않은 평생의 고통으로 이어질 수 있다는 점에서 그저 ‘스캔들’로 부르기에는 부족해 보인다. 게다가 새로운 의료기기들이 대부분 매우 비싸다는 점, 신기술의 안전성과 유효성에 대한 근거가 충분하지 않은 상황에서 빠르게 허가를 내주고 건강보험 적용을 받게 해주려는 현 정부의 기조를 생각하면 문제는 더욱 심각하다. 이는 세금과 보험료로 충당된 건강보험 재정이 분배정의에 합당하게 사용되고 있는지에 대한 문제이면서, 동시에 건강과 생명에 대한 중대한 위협이기도 한 셈이다.
지난 2014년 미국 국립건강연구센터(National Center for Health Research)에 소속된 세 명의 연구자가 <미국의사협회 내과학회지(JAMA Internal Medicine)>에 발표한 논문은 바로 이러한 문제를 다루고 있다. (☞관련 논문 : 인체이식형 의료기기의 안전성과 유효성에 대한 공개된 과학적 정보의 부재)
미국에서 의료기기 허가의 대부분은 기존에 출시된 제품과 상당히 비슷하다는 사실만 입증하면 안전성과 유효성에 대한 별도의 과학적 근거 없이도 시판 허가를 내어주는 510(k) 절차를 통해 이루어진다. 이 논문에 따르면 이러한 약식 절차가 아닌 정식 절차를 거쳐 허가받는 제품은 매년 채 20개가 되지 않는다. 이러한 의료기기 시판 허가 절차는 안전을 충분히 보장할 수 있을까? 유감스럽게도, 그렇지 않은 것 같다. 예를 들어 새로운 의료기기 A가 기존 시판 제품 B와 동등하다는 것을 인정받아 시판허가를 받았다고 해보자. 이후 B의 안전 문제가 발견되어 회수조치를 당하는 경우에도, 의료기기 A의 허가에는 영향을 미치지 않는다. 동등함을 근거로 허가받았지만, 막상 문제가 발견되었을 때는 별도의 제품처럼 취급되는 것이다. 또다른 문제도 있다. 이를테면 금속과 금속이 맞닿도록 만들어진 고관절 치환물의 경우, 기존 제품과 동등하다는 인정을 받아 출시되지만 이들 각각은 완전히 똑같은 제품이 아니라 조금씩 개량과 변형을 거듭한 것들이다. 따라서 각기 다른 안전성 문제를 갖게 될 가능성이 있고, 출시 제품의 종류가 많을수록 이런 문제는 더 커진다. 연구진은 이런 이유들 때문에 개별 의료기기마다 적절한 과학적 근거로 뒷받침되는 것이 매우 중요하다고 주장하며, 논문에서 다음의 세 가지를 확인하고자 했다.
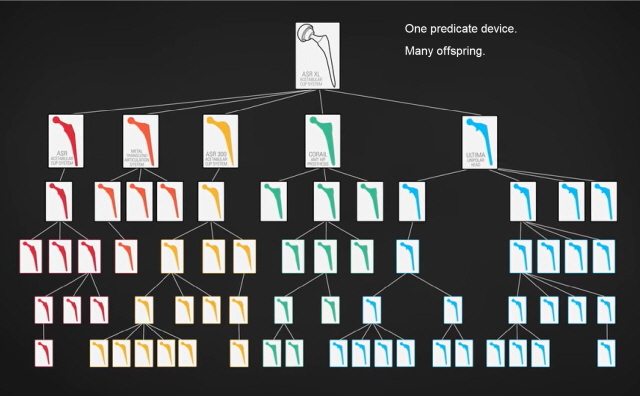
첫째, 신체에 이식하는 의료기기의 대표성 있는 표본을 추출했을 때 이들이 기존 제품과 상당히 비슷한지, 안전한지, 효과적인지를 결정하는데 사용된 과학적 근거에는 어떤 것이 있는가?
둘째, 허가 당시에 해당 제품과 유사한 기존의 의료기기는 몇 종류나 있었는가?
셋째, 이런 근거들이 대중에 공개되어 있는가?
연구진은 이 질문에 답하기 위해 미국 FDA 자료로부터 2008년 510(k) 절차를 통해 허가받은 인체이식형 의료기기들을 분석 대상으로 삼았다. 대분류 체계 상 심혈관계, 치과계, 외과계, 신경계, 정형외과계로 분류된 기기들 중, 2008~2012년 동안 가장 먼저 허가받은 인체이식형 의료기기를 매년 두 개씩 뽑아 총 50개의 의료기기를 선정했다. 그리고 이들 의료기기가 허가받는데 이용된 1105개의 기존 의료기기들을 확인했다. 이어 연구진은 확인한 의료기기들이 상당히 비슷하다는 주장을 뒷받침하는 과학적 근거가 FDA에 제출되었는지, 제출된 자료가 공개되어 있는지 확인하고, 안전성과 유효성에 대한 자료가 제출되었는지를 추가로 검토했다.
확인 결과, 새롭게 허가된 50개 의료기기 중 8개만이 기존 기기와 상당히 유사하다는 과학적 근거를 제출한 것으로 나타났다. 임상적 근거와 비임상적 근거 모두를 제출하고 허가받은 기기는 단 한 개 뿐이었다. 최근에 허가받은 기기일수록 더 많은 근거를 제출했는지 검토했지만, 최근 허가받은 10개 의료기기 중 2개만이 과학적 근거를 명시한 것으로 드러났다. 허가의 근거가 된 기존 의료기기들도 전체 1105개 중 31개만이 과학적 근거를 제출하고 허가를 받았으며, 요약 정보가 없었던 602개 중 최소 264개 이상이 근거 제출을 요구하는 안전 의료기기 법(Safe Medical Devices Act)이 시행되기 전에 허가받은 제품이었다.
연구진은 의료기기가 안전하고 효과적이라는 근거가 충분하지 않으며, 또한 동등성, 안전성, 유효성에 대한 과학적 근거가 법이 요구하는 수준으로 공개되지 않았다고 결론 내렸다. 그러면서 시민의 건강을 지키고, 의료기기의 효과에 대한 과학적 근거를 공정하게 평가하기 위해 FDA가 의료기기 시판허가와 관련된 법을 강화해야 한다고 촉구했다.
한국에서는 아직 이와 비슷한 연구가 수행되지 않았지만, 한국 식품의약품안전처의 의료기기 허가 절차가 미국 FDA의 510(k) 절차를 표본으로 삼고 있다는 점에서 상황은 대체로 비슷할 것으로 짐작된다. 또한 유럽의약품기구가 허가한 항암제의 대부분이 전체 생존율이나 삶의 질을 향상시킨다는 근거가 없고, 근거가 있더라도 그 효과가 제한적이라는 2017년 <영국의사협회지 British Medical Journal> 논문을 본다면, 이러한 문제는 단지 의료기기에 국한되지 않을 것이다. (☞관련 논문 : 유럽의약품기구 승인을 받은 항암제의 전체 생존율과 삶의 질에 대한 혜택 근거의 가용성: 2009-2013에 이루어진 약품 허가에 대한 후향적 코호트 연구).
시민들은 정부의 허가 절차를 통과한 의약품, 의료기기이니 당연히 과학적 효과성과 안전성이 입증되었을 것이라고 생각한다. 그러나 오늘 소개한 논문은 현실이 그렇지 않음을 보여준다. 한정된 공적 자원의 적절한 활용을 위해서, 또 무엇보다도 시민들의 건강과 안전을 지키기 위해서도, 정부는 규제와 감시의 역할을 강화해야 할 것이다. 신성장동력을 꿈꾸며 규제완화를 이야기할 할 것이 아니라, 시민의 건강과 안전 보호라는 가장 기본적인 책무를 다하기 위한 노력이 필요하다. (☞관련 글 : 신의료기술의 규제완화, 누구를 위한 것인가?)